Chorea Huntington - Entstehung und Diagnose
Bei Chorea Huntington handelt es sich um eine Erbkrankheit, die unausweichlich tödlich verläuft. Die betroffenen Personen erkranken durchschnittlich mit dem 40igsten Lebensjahr. Die Krankeit ist gekennzeichnet durch Bewegungsstörungen und schwere psychische Veränderungen.
Worum gehts?
Wie wird die Krankheit Chorea Huntington vererbt und wie lässt sie sich diagnostizieren?
Vererbung von Chorea Huntington
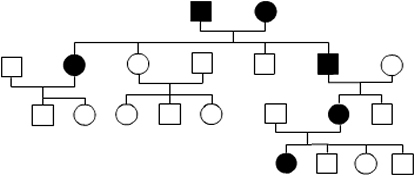
Erbkrankheiten des Menschen können rezessiv bzw. dominant und vom Geschlecht abhängig vererbt werden. Will man die Vererbung von Erbkrankheiten klären, so untersucht man Familien, in denen die Krankheit über mehrere Generationen auftritt. Das Ergebnis wird in Familienstammbäumen dargestellt. Die Auswertung von Stammbäumen gibt Aufschluss über den Vererbungsmechanismus.
- Anhand des Stammbaumes wird deutlich, dass Chorea dominant vererbt wird. Das betroffene Elternpaar hat gesunde Kinder. Das ist nur möglich, wenn beide Eltern ein gesundes Gen weitergeben.
- Die Krankeit wird autosomal vererbt. Es sind Männer und Frau gleichmäßig betroffen. Außerdem hat der kranke Vater eine gesunde Tochter. Das wäre nicht der Fall, wenn das Gen auf dem X-Chromosom des Vaters liegen würde. Das Huntingtin-Gen liegt somit nicht auf dem X-Chromosom.
Molekulargenetische Ursache von Chorea Huntington
- Die Erbkrankheit Chorea Huntington wird durch Genmutation im Huntingtin - Gen verursacht.
- Das Gen befindet sich auf dem Chromosom 4.
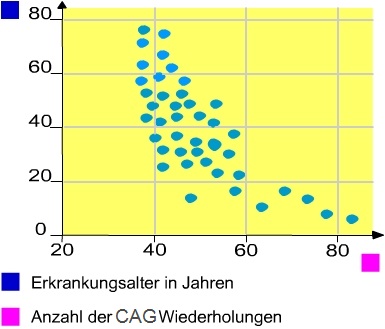
- Im codierenden Strang können zwischen 5 und 30 CAG Tripletts („short tandem repeats“) aufeinander folgen.
- Eine Erhöhung der Tripletts führt zum Ausbruch der Huntingtonschen Krankheit.
- Zusätzlich ist die Zahl der Wiederholungen über 30 instabil und steigt tendenziell mit jeder Generation (Replikation).
- Die CAG Tripletts codieren für die Aminosäure Glutamin. Die Erhöhung führt zu einer Verlängerung der Glutaminkette im Protein.
- Ab einer bestimmten Länge verklumpen die Proteine, wodurch schließlich Nervenzellen absterben.
Isolation des Huntingtin – Gens durch PCR
- Die molekulargenetische Diagnostik von Chorea Huntington erfolgt über die Längenbestimmung von spezifisch verdoppelten PCR-Produkten und der sich daraus ableitenden Anzahl an CAG-Triplett-Wiederholungen.
- Die genaue Längenbestimmung der gewonnen DNA-Schnipsel erfolgt mittels der Gelelektrophorese (siehe weiter unten).
- Nicht verlängerte und verlängerte Genabschnitte werden durch mehrere PCR-Zyklen mit unterschiedlichen Primerpaaren zu den notwendigen Mengen vervielfacht.
- Voraussetzung für die Isolation des Huntingtin - Gens sind spezifische Primer, die sich an den Erkennungssequenzen (ES) anlagern.
Darstellung des Huntingtin - Gens
Die in der PCR isolierten Gen-Abschnitte werden nun mit Hilfe der Gelelektrophorese aufgetrennt. Anhand der Ergebnisse lässt sich feststellen, welche Person Träger des defekten Huntingtin-Gens ist.
- Die Schnelligkeit mit der die isolierten Gene das Gel durchlaufen ist abhängig von ihrer Länge.
- Kurze Gene (DNA - Abschnitte) sind scheller als lange.
- Der Marker M18 enthält 18 CAG - Tripletts, der Marker M48 dagegen die angegebene Anzahl von 48 CAG - Tripletts.
- Mit den Markern lassen sich die Genlängen der zu untersuchenden Personen schätzen.
Auswertung
- Beide Eltern verfügen über ein gesundes und ein krankes Gen. Welches sie an die Kinder weitergeben ist zufällig. Das defekte Gen des Vaters ist länger als das der Mutter.
- Das Kind 1 (K1) ist reinerbig für die gesunden Gene. Es wird nicht erkranken.
- Die Kinder 2 (K2) und 4 sind reinerbig für die kranken Gene. Sie werden erkranken.
- Mischerbig sind die Kinder 3 und 5. Sie werden ebenfalls erkranken.
- Bei den Kinder 4 und 5 sind die kranken Gene länger als bei den Eltern. Man könnte meinen, dass ihr defektes Gen nicht vom Vater stammen kann. Aber leider ist es so, dass es während der Replikation zu einer Genverlängerung kommen kann.
...oder ein Feedback abgeben.>>>>>>>>